Bệnh cơ tim phì đại (BCTPĐ) là 1 bệnh tim mạch được đặc trưng bởi rối loạn sự phát triển các sợi tơ cơ của cơ tim dẫn đến phì đại bất thường cơ thất trái. Sự phì đại này có thể ở thành tâm thất trái (đồng tâm) hoặc ở vách liên thất (lệch tâm). Tỷ lệ mắc bệnh ước tính khoảng 1/500-1000 người. Bệnh nhân bị BCTPĐ có nguy cơ bị các rối loạn nhịp tim như tim nhanh thất hay rung thất, có thể dẫn đến đột tử mà không có dấu hiệu báo trước. Trên các bệnh nhân BCTPĐ hoặc nghi ngờ BCTPĐ có tính chất gia đình, 50-60% trường hợp tìm thấy đột biến một trong các gen mã hoá các thành phần tạo ra sợi tơ cơ. Mục tiêu: Phát hiện đột biến gen MYH7 của bệnh nhân mắc cơ tim phì đại. Đối tượng nghiên cứu: Bệnh nhân nam, 36 tuổi, chẩn đoán BCTPĐ, có cơn choáng ngất thoảng qua. Phương pháp nghiên cứu: Phân tích gen MYH7 bằng kỹ thuật di truyền phân tử. Kết quả: Phát hiện đột biến Arg403Glu tại exon 13 của gen MYH7. Kết luận: Nhân trường hợp phát hiện đột biến Arg403Glu gen MYH7 ở bệnh nhân BCTPĐ đã làm sáng tỏ thêm về cơ chế BCTPĐ tại Việt Nam, mở thêm hướng chẩn đoán đối với BCTPĐ, giúp người thầy thuốc tầm soát bệnh tốt hơn.
Từ khóa: MYH7, Cơ tim phì đại. SUMMARY
DETECTION OF MUTATION IN THE BETA - MYOSIN HEAVY CHAIN (MYH7) IN HYPERTROPHIC CARDIOMYOPATHY
Hypertrophic cardiomyopathy (HC) is a cardiac disease characterized by a sarcomeric disarray that leads to cardiac muscle cell hypertrophy. The prevalence of HC has been estimated in 1 in 500 to 1000 persons, and HC is the most common cause of sudden death in the young. The genes involved in HC encode proteins of the sarcomere. The first HC locus was mapped to the long arm of chromosome 14 (14q1). The gene encodes the beta-myosin heavy chain (MYH7) gene, and more than 50 different mutations have been identified worldwide. Objectives: To investigate mutation on MYH7 gene. Subject: A 36 year-old male patient, history of hypertrophic cardiomyopathy. Methods: sequencing MYH7 gene, using reference and control. Results: The mutation Arg403Glu of MYH7 gene was found in HC patient. Conclusion: Our study illustrates that detection MYH7 gene mutations in HC patient has shed light on the pathogenesis of HC in Vietnam, more open diagnosis direction for HC and help doctors screening better for this disease.
Key words: MYH7 gene, Hypertrophic cardiomyopathy. I. ĐẶT VẤN ĐỀ Bệnh cơ tim phì đại (BCTPĐ) là 1 bệnh tim mạch được đặc trưng bởi rối loạn sự phát triển các sợi tơ cơ của cơ tim dẫn đến phì đại bất thường cơ thất trái. Sự phì đại này có thể ở thành tâm thất trái (đồng tâm) hoặc ở vách liên thất (lệch tâm). Tỷ lệ mắc bệnh ước tính khoảng 1/500-1000 người [1]. Bệnh nhân bị BCTPĐ có nguy cơ bị các rối loạn nhịp tim như tim nhanh thất hay rung thất, có thể dẫn đến đột tử mà không có dấu hiệu báo trước. Đây là một nguyên nhân hàng đầu dẫn đến đột tử ở các bệnh nhân dưới 35 tuổi [16]. Sự phì đại thất trái hay gặp nhất là phì đại vách liên thất, chiếm tới 46% [9], có thể có hay không kèm theo sự tắc nghẽn đường ra thất trái. Lâm sàng của bệnh nghèo nàn và thay đổi tuỳ thuộc vào tuổi phát hiện bệnh, mức độ nặng nhẹ của quá trình tiến triển bệnh. BCTPĐ có thể biểu hiện ở trẻ nhỏ mặc dù người ta thường phát hiện bệnh ở tuổi thiếu niên hoặc ở người trưởng thành [16], [9]. Nh÷ng quan t©m gÇn ®©y vÒ gen di truyÒn cña bÖnh lý này ®· më ra nhiều híng míi trong tiªn lîng, theo dâi và ®iÒu trÞ.
Trên các bệnh nhân BCTPĐ hoặc nghi ngờ BCTPĐ có tính chất gia đình, 50-60% trường hợp tìm thấy đột biến một trong các gen mã hoá các thành phần tạo ra sợi tơ cơ. BCTPĐ được xác định tuân theo quy luật di truyền trội trên nhiễm sắc thể thường. Các nhà khoa học đã tìm thấy trên 900 đột biến thuộc 20 gen liên quan đến bệnh. Tất cả các gen đều tham gia mã hóa cấu trúc cho các protein của tế bào cơ tim. Thành phần chủ yếu của cơ tim là xơ actin và xơ myosin. Xơ myosin sắp xếp với xơ actin thành đơn vị co duỗi cơ. Trên mỗi đơn vị co duỗi cơ myosin - actin còn có gắn thêm các phân tử troponin gồm 3 tiểu đơn vị là A, C và T. Các đột biến gây BCTPĐ thường liên quan đến gen tổng hợp protein cấu tạo xơ myosin (MYH7) và troponin (TNNT, TPMA, MYBPC, TNNI3…), trong số này, đột biến gen MYH7 chiếm tới 30 - 45% các trường hợp. [16], [9], [21].
Phân tích gen đột biến cho phép phát hiện và chẩn đoán những trường hợp có nguy cơ cao bị BCTPĐ trước khi có biểu hiện lâm sàng. Có đột biến gen MYH7 khẳng định chắc chắn BCTPĐ khi có các triệu chứng lâm sàng. Nếu đột biến gen được phát hiện thấy ở những bệnh nhân chưa có biểu hiện lâm sàng thì những trường hợp này bắt buộc phải theo dõi định kỳ.
Ở Việt Nam, lần đầu tiên chúng tôi tiến hành nghiên cứu gen MYH7 ở các bệnh nhân BCTPĐ với mong muốn tìm hiểu mối liên quan tới việc biến đổi kiểu gen và đặc điểm lâm sàng của BCTPĐ, đồng thời dự đoán trước nguy cơ mắc bệnh cũng như tư vấn dự phòng biến cố tim mạch như rối loạn nhịp, ngất, đột tử, khả năng di truyền bệnh,…Chúng tôi xin báo cáo một trường hợp BCTPĐ đã phát hiện thấy có đột biến gen MYH7. II. ĐỐI TƯỢNG VÀ PHƯƠNG PHÁP NGHIÊN CỨU 2.1. Đối tượng nghiên cứu: Bệnh nhân nam, 36 tuổi, nhập viện ngày 05/9/2011 vì có các cơn choáng và đau thắt ngực.
Bệnh nhân có tiền sử đã được chẩn đoán BCTPĐ từ 2003, nhưng điều trị không đều. Nhưng đến 2007 ngoài đau ngực, bệnh nhân thấy khó thở khi gắng sức. Vì vậy, bệnh nhân đã chủ động đi khám và được theo dõi định kỳ và điều trị đều hàng tháng (tildiem 60 mg uống 2 viên/1 ngày). Khoảng mấy tháng trước khi nhập viện, bệnh nhân xuất hiện các cơn choáng thoáng qua.
Trong gia đình bệnh nhân: Mẹ, 2 chị và anh trai cũng được chẩn đoán BCTPĐ.
Bệnh nhân được thăm khám và siêu âm Doppler tim và các thăm dò chức năng khác để chẩn đoán xác định và đánh giá mức độ BCTPĐ. Bệnh nhân được lấy máu làm các xét nghiệm cận lâm sàng, đồng thời phân tích gen MYH7 tại Bộ môn Y sinh học - Di truyền, trường Đại học Y Hà Nội. 2.2. Phương pháp nghiên cứu:
2.2.1. Thăm khám lâm sàng.
2.2.2. Chẩn đoán hình ảnh: Chụp tim phổi thẳng.
Siêu âm Doppler tim.
Thông tim ống lớn và Chụp động mạch vành: đánh giá mức chênh áp ở đường ra thất trái, mức độ hở van hai lá, hệ động mạch vành: xét đốt vách liên thất. 2.2.3. Xét nghiệm cận lâm sàng:
Các xét nghiệm : công thức máu, sinh hóa máu
Điện tâm đồ. 2.2.4. Phân tích gen MYH7 DNA được tách chiết từ tế bào bạch cầu lympho máu ngoại vi bằng kit Phenol/ChCl3. Các exon chứa các đột biến có nguy cơ gây BCTPĐ (exon 13 và 14) được nhân lên bằng phản ứng PCR với 40 chu kì luận nhiệt (95oC-5 phút/55oC-30 giây/72oC-30 giây). Thể tích phản ứng 50 µl, bao gồm các thành phần: DNA mẫu, mồi, và các thành phần cho phản ứng PCR (Taq DNA polymerase, dNTP, MgCl2, dH2O).
Trình tự mồi sử dụng cho exon 13-14: Mồi xuôi: 5’-CAG GCA TGA ACC ACA CAC CTG - 3’) Mồi ngược: 5’- TCT CAT CCC ACC ATG CCA GT-3’. Đoạn gen MYH7 được nhân lên có kích thước 488 bp.
Sản phẩm PCR được tinh sạch bằng kit PureLinkTM (Invitrogen). Xác định trình tự gen FGB được thực hiện trên máy ABI PRISM 3100 Genetic Analyzer. Các thông số và chất lượng đỉnh được thu thập, kiểm định bằng các phần mềm ABI Data Collection v2.0 và Sequencing Analysis Sotwave v5.3. Trình tự đoạn gen MYH7 được so sánh với trình tự tham chiếu công bố trên GenBank thông qua sử dụng phần mềm phân tích Chromas Lite v2.1.1 và Seaview để xác định đột biến. III. KẾT QUẢ NGHIÊN CỨU
3.1. Đặc điểm lâm sàng và cận lâm sàng của bệnh nhân Lúc nhập viện, bệnh nhân tỉnh táo, đau ngực trái không rõ ràng, không khó thở, tần số tim 120 ck/phút, huyết áp 2 tay 120/70 mmHg, hai phổi không có rale, gan không to. Điện tâm đồ: Vẫn là nhịp xoang đều 120 ck/ph, trục xu hướng trái, dày thất trái, ST chênh xuống nhẹ ở các chuyển đạo D2, aVF, V5-6. Các xét nghiệm công thức máu, đông máu, sinh hoá máu: đều trong giới hạn bình thường. Không có bằng chứng của: nhồi máu cơ tim, suy tim, tiểu đường. Siêu âm Doppler tim: Hình ảnh BCTPĐ lệch tâm, chức năng tâm thu thất trái bình thường, giảm chức năng tâm trương thất trái, tăng áp lực đổ đầy thất trái, hở hai lá nhẹ. Cụ thể như sau: đường kính thất trái cuối tâm trương Dd =35 mm; đường kính thất trái cuối tâm thu Ds = 24 mm; thể tích thất trái cuối tâm trương Vd = 50 ml; thể tích thất trái cuối tâm thu Vs = 20,6 mm; các thành thất trái dày (tâm trương 12 mm, tâm thu 13 mm); phì đại toàn bộ vách liên thất (tâm trương 23 mm; tâm thu 24 mm), có dấu hiệu SAM; hở hai nhẹ (diện tích hở hai lá 2,4 cm2); không có tăng chênh áp ở đường ra thất trái (max 8 mmHg, trung bình 4 mmHg); E/A = 74/54; E/E’ = 24,66; Dt = 243 ms. Thông tim ống lớn và chụp động mạch vành kiểm tra: Không thấy có tăng chênh áp ở đường ra thất trái; hệ động mạch vành bình thường. Bệnh nhân được điều trị thuốc: Metoprolol (Betaloc zok 50 mg/1 ngày) và ra viện 03 ngày sau đó (08/9-2011). 3.2. Đặc điểm gen MYH7 của bệnh nhân
3.2.1. Nhân gen MYH7 bằng kỹ thuật PCR
DNA tổng số được chiết tách dùng làm khuôn để nhân các đoạn exon 13 - 14

Hình 3.1. Điện di sản phẩm PCR của bệnh nhân
M: Thang DNA chuẩn 100bp
Kết quả điện di sản phẩm PCR (Hình 3.1) cho thấy sản phẩm PCR đặc hiệu và rõ nét ở mẫu nghiên cứu, gen MYH7 có kích thước 488bp, phù hợp với thiết kế nghiên cứu.
3.2. Trình tự gen MYH7 của bệnh nhân
Sản phẩm PCR sau khi được tinh sạch được tiến hành xác định trình tự theo cả hai chiều (chiều xuôi - F và chiều ngược - R) của gen MYH7.
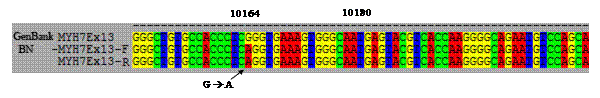
Hình 3.2. Trình tự nucleotide đoạn gen MYH7 của bệnh nhân
So sánh với trình tự tham chiếu tại ngân hàng gen (GenBank), chúng tôi phát hiện trên exon 13 có đột biến sai nghĩa do thay thế nucleotide loại Guanine (G) thành nucleotide loại Adenine (A) tại vị trí 10164, dẫn đến làm thay đổi bộ ba mã (codon) CGG thành CAG hay làm thay đổi axit amin Arginine thành Glutamine tại vị trí 403 (Arg403Glu hay R403Q).
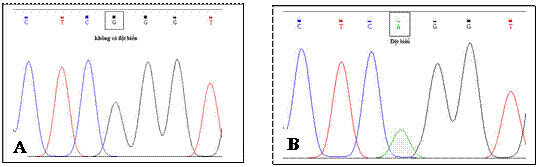
Hình 3.3. Đỉnh các điểm nucleotide trên vùng gen MYH7 của bệnh nhân
A: Gen MYH7 người bình thường không có đột biến; B: Gen MYH7 của bệnh nhân bị đột biến GàA tại vị trí 10164. IV. BÀN LUẬN Chẩn đoán xác định BCTPĐ trên bệnh nhân này là hoàn toàn rõ ràng dựa trên các yếu tố: siêu âm Dopler tim có bề dày vách liên thất tâm trương 23 mm, tâm thu 24 mm. Theo tài liệu đồng thuận về guideline cho BCTPĐ giữa ACC, ESC (2003) khi thành tâm thất trái bao gồm cả vách liên thất dày lớn hơn hoặc bằng 15 mm thì được chẩn đoán là bệnh cơ tim phì đại [13]. Chẩn đoán thể BCTPĐ trên bệnh nhân này là thể lệch tâm, không có tăng chênh áp ở đường ra thất trái: các thành thất trái dày nhẹ (tâm trương 12 mm, tâm thu 13 mm); chênh áp ở đường ra thất trái (max 8 mmHg, trung bình 4 mmHg).
SAM là hình ảnh bất thường vận động đặc trưng nhất của BCTPĐ. Hình ảnh này tương ứng với vận động bất thường toàn bộ van hai l¸ về phÝa v¸ch liªn thất trong thêi t©m thu ®îc gi¶i thÝch do nâng lên quá mức các cơ nhó v× thành trước và thành sau tăng cường vận động hậu quả là làm trùng lại d©y chằng van hai l¸[13].
Triệu chứng cơ năng của BCTPĐ thưòng nghèo nàn. Ba triệu chứng kinh điển thường gặp trong BCTPĐ là : khó thở, đau thắt ngực và ngất [18,26]. Triệu chứng khó thở thường gặp nhất , chiếm đễn 90% các trường hợp, được giải thích do rối loạn chức năng tâm trương của thất trái [28]. Mức độ khó thở không có liên quan gì tới tình trạng huyết động, hoặc mức độ chênh áp của đường ra thất trái cũng như chức năng tâm thu thất trái [15]. Đau thắt ngực chiếm 70-80% các trường hợp BCTPĐ [3] và 15 % có phối hợp nhồi máu cơ tim [10]. Đau ngực không do nhồi máu cơ tim được giải thích là do rối loạn chức năng tâm trương thất trái dẫn đến mất cân bằng cung cầu oxy của cơ tim gây so thắt động mạch vành [17,28].
Ngất chiếm 20% BCTPĐ, nhưng 50-55% trong số này thường có triệu chứng báo trước [15], thường có liên quan tới gắng sức vì khi gắng sức làm tăng đáng kể chênh áp ở đường ra thất trái [17]. Tuy nhiên, nhiều nghiên cứu lại cho thấy ngất lại xảy ra ngay lúc nghỉ ngơi và không có tăng chênh áp ở đường ra thất trái. Những trường hợp này được giải thích là do rối loạn nhịp, phổ biến là rối loạn nhịp thất [4,25].
Bệnh cơ tim phì ®¹i chủ yếu là do đột biến trong một trong những gen quy định tổng hợp tế bào cơ tim (sarcomeric gene). Cho đến nay, khoảng 194 đột biến đã được xác định ở người bị bệnh cơ tim phì đại, trong số đó 115 đột biến xảy ra trên gen MYH7 [8]. Gen MYH7 gồm có 39 exon và nằm trên nhánh dài nhiễm sắc thể số 14 (14q12). Gen MYH7 mã hóa cho chuỗi nặng b myosin. Đột biến gen MYH7 được cho là nguyên nhân thường gặp nhất trong BCTPĐ. Có 50 loại đột biến trên MYH7 đã được xác định, phần lớn các đột biến nằm trong các vùng mã hóa từ exon 8 - 24. Các vùng này mã hóa tổng hợp protein tạo khối cầu myosin trong việc kết hợp với xơ actin hình thành đơn vị co cơ.
Một số nghiên cứu về vai trò của đột biến gen MYH7 trong sự phát triển của bệnh cơ tim phì đại, chẳng hạn như Wang và cs. (2008) đã thu thập mẫu máu của hai gia đình mắc bệnh cơ tim phì đại và 120 người khỏe mạnh để nghiên cứu đột biến gen ở các gen MYH7, MYBPC3, và TNNT2 bằng phương pháp PCR và xác định hầu hết những đột biến xảy ra đều ở trên gen MYH7 [30]. Pare và cs. (1961) đã chứng minh mối liên quan của các rối loạn về tim (192.600) ở nhóm người Canada gốc Pháp và vùng gen thuộc nhánh dài nhiễm sắc thể 14, nơi chứa gen MYH7 [22]. Garcia và cs. (Úc) giải trình tự exon 13 - 16 của gen MYH7 trong 30 bệnh nhân cơ tim phì đại, trong độ tuổi từ 18 - 60, trong đó có 25 bệnh nhân có tiền sử gia đình mắc bệnh cơ tim phì đại. Trong nghiên cứu này phát hiện ở 1 bệnh nhận xuất hiện 1 đột biến (Arg453Cys, exon 14) trên gen MYH7 [6]. Villard và cs. đã phân tích tất cả các exon của hai gen MYH7 và TNNT2 sử dụng phương pháp SSCP ở 96 bệnh nhân (54 bệnh nhân có tiền sử gia đình và 42 bệnh nhân không thường xuyên mắc bệnh ). Đột biến gen MYH7 đã được xác định ở 5 bệnh nhân có tiền sử gia đình và ở 2 bệnh nhân không thường xuyên mắc bệnh. Trong nghiên cứu này đã phát hiện ra rằng hầu hết các đột biến đều ở gen MYH7 và cũng là thời điểm khởi phát bệnh bị chậm lại. Garcia và cs. nghiên cứu tần số của gen sarcomere ở 120 bệnh nhân cơ tim phì đại tại Tây Ban Nha, trong số đó, 16% có tiền sử gia đình. Họ đã nghiên cứu kiểm tra trên 5 gen và xác định được các đột biến ở 32 bệnh nhân, trong đó có 10 đột biến trên gen MYH7 [5]. Tại Iran, Montazeri và cs. đã nghiên cứu các đột biến phổ biến trên gen MYH7, bao gồm exon 13-15 ở 50 bệnh nhân và xác định được đột biến G10195A trong exon 13 và một đột biến A10419C trong exon 14 [20]. Các kết quả của một vài nghiên cứu ở Iran và nghiên cứu sâu rộng trong các phần khác của thế giới về gen này với sự ảnh hưởng của chúng đến bệnh cơ tim phì đại cho rằng exon 8-24 của gen MYH7 ảnh hưởng nhiều nhất trong căn bệnh này. Một số đột biến thuộc gen MYH7 được cho là có ý nghĩa tiên lượng bởi liên quan đến nguy cơ đột tử của bệnh như Arg403Gln, Arg719Trp, Arg453Cys [1,31]. Tuy nhiên, một số trường hợp đột biến chỉ là người mang gen, không biểu hiện bệnh như: Gly256Glu, Val606Met, Leu908Val [1,29].
Nghiên cứu của Liu và cộng sự ®Ó sàng lọc các đột biến của gen có nguy cơ gây bÖnh cơ tim phì đại trong 10 gia hệ ở Trung Quốc. Trong nghiên cứu này, trong 3 gia hệ đã xác định có đột biến gen MYH7, 3 bệnh nhân đã đột ngột qua đời ở tuổi 20 - 48 trong khi tập luyện thể thao. Kết quả của nghiên cứu này cho thấy các đột biến của gen MYH7 ở những bệnh nhân cơ tim phì đại có liên quan với nguy cơ đột tử cao do tim [12]. Trong một gia đình Trung Quốc gồm 3 thế hệ, Ko và cs. (1996) đã quan sát thấy sự cùng tồn tại của đột tử và suy tim giai đoạn cuối do đột biến Arg453Cys [11]. Tuổi trung bình của các thành viên bị đột tử có tính chất gia đình là 34 năm. Geisterfer-Lowrance và cs. (1990) tìm thấy một đột biến sai nghĩa trong chuỗi beta myosin ở bệnh nhân cơ tim phì đại dẫn đến làm thay thế arginine thành glutamine tại vị trí 403 (R403Q) [7]. Perryman và cs. (1992) chứng minh rằng đột biến R403Q thường gặp trên phân tử mRNA của cơ tim [23]. Radovan và cs. mô tả trường hợp trẻ tuổi BCTPĐ tắc nghẽn mang đột biến Arg403Glu tại exon 13 của gen MYH7 [24]. Spindler và cs. (1998) đã nghiên cứu ảnh hưởng của đột biến tại codon 403 của gen MYH7. Họ đã quan sát thấy 3 sự thay đổi lớn trong sinh lý học và năng lượng sinh học ở tim chuột đột biến. Đầu tiên, trong khi không bị rối loạn chức năng tâm thu, chức năng tâm trương bị suy giảm trong thời gian kích thích co bóp . Thứ hai, trong điều kiện chuột bị đột biến R403Q thì tim chuột có hàm lượng phosphocreatine thấp hơn và tăng hàm lượng phosphate vô cơ dẫn đến giảm giá trị năng lượng tự do sinh ra từ quá trình thủy phân ATP. Thứ ba, chuột bị đột biến tim đã được tăng perfusate canxi bằng cách giảm xuống 2 lần nhịp tim so với đối chứng. Các tác giả trên đã kết luận rằng tim của chuột mang đột biến R403Q đã bị phụ thuộc vào việc làm rối loạn chức năng tâm trương tương tự như biểu hiện của người bị bệnh cơ tim phì đại có tính chất gia đình [27].
Trong nghiên cứu của chúng tôi, gen MYH7 đã được kiểm tra và xác định được trên exon 13 xuất hiện đột biến sai nghĩa do thay thế nucleotide loại Guanine (G) thành nucleotide loại Adenine (A) tại vị trí 10162, dẫn đến làm thay đổi codon CGG thành CAG hay làm thay đổi axit amin Arginine thành Glutamine tại vị trí 403 (Arg403Glu hay R403Q). Việc phát hiện này giúp chúng tôi lý giải rõ hơn về biểu hiện lâm sàng của bệnh nhân, với những cơn choáng ngất thoảng qua là một trong các triệu chứng của BCTPĐ và là tiền triệu của cái chết đột tử dễ xảy đến với nhóm bệnh nhân mang loại đột biến này. Đồng thời dựa vào vị trí và kiểu đột biến gen có thể giúp tiên lượng kiểu hình cũng như mức độ nặng (nguy cơ rối loạn nhịp dẫn đến đột tử ) những trường hợp BCTPĐ. V× vËy, nếu test gen MHY7 âm tính và không có triệu chứng lâm sàng thì những trường hợp này được khẳng định chắc chắn nguy cơ mắc BCTPĐ là rất thấp mặc dù họ được sinh ra trong gia đình có tiền sử bị BCTPĐ mang đột biến gen MYH7.
V. KẾT LUẬN Lần đầu tiện tại Việt Nam, người bệnh BCTPĐ được phân tích gen MYH7 và phát hiện được đột biến có liên quan tới biểu hiện bệnh. Mặc dù đây không phải là đột biến mới và đã được thế giới công bố, nghiên cứu của chúng tôi cho thấy đã làm sáng tỏ thêm về cơ chế BCTPĐ tại Việt Nam, mở thêm hướng chẩn đoán đối với BCTPĐ, giúp người thầy thuốc tầm soát bệnh tốt hơn. |